



根据欧盟医疗器械法规(MDR 2017/745)*120条,新增型号不一定需要重新申请CE认证,但必须严格评估变更性质。关键判定依据包括:
| 变更类型 | 典型案例 | 合规要求 |
|---|---|---|
| 技术文档更新 (*公告机构介入) | • 外观颜色调整 • 非关键部件供应商变更 | 更新技术文件, 内部评审记录 |
| 重大变更 (需公告机构审查) | • 新增适应症 • 核心算法修改 • 灭菌方式改变 | 提交变更申请, 可能需补充临床评估 |
| 全新产品 (需重新认证) | • 工作原理改变 • 风险等级提升 | 全新MDR认证流程 |
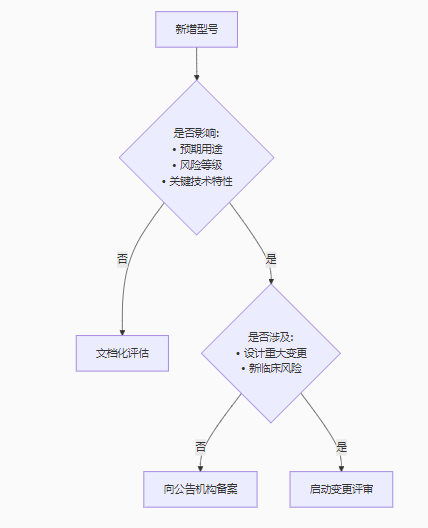
非灭菌/非测量类:可自主更新技术文件
灭菌/测量类:需公告机构审核关键变更
材料变更、软件重大升级需公告机构批准
典型审核周期:4-8周
任何可能影响临床性能的变更均需:
补充临床数据
小组咨询(如适用)
平均处理时间:3-6个月
产品分类变化(如IIa→IIb)
新增预期医疗用途(如诊断→*)
核心技术创新(如新增AI功能)
同系列扩展:通过MEDDEV 2.1/1 Rev.4评估
参数调整:提供等效性验证报告
配件兼容:更新技术文件附录
针对型号扩展需求,我们提供全流程合规解决方案:
✅ 变更影响评估:72小时出具法律意见书
✅ 技术文件升级:符合MDR Annex II/III要求
✅ 公告机构沟通:加速变更评审流程(合作NB包括TÜV SÜD、BSI等)
✅ 临床评估补充:按MEDDEV 2.7/1 Rev.4执行
✅ 上市后监督:更新PMS/PMCF计划
2024年成功案例:
协助某骨科企业3周内完成:
5个新型号植入物的同系列认证扩展
节省60%认证成本
建立自动化变更管控系统
欧盟MDR监管日趋严格,专业支持决定市场准入效率!
立即联系角宿MDR团队,获取定制化变更方案。
FDA,监督,已上市器械
当一款医疗器械成功获得FDA批准或许可并上市后,许多企业可能会认为主要的合规工作已经完成。然而,事实恰恰相反,产品的上市标志着企业进入了一个新的、至关重要的阶段——上市后监管。FDA建立了一套严密、主动的持续监督体系,以确保在美国市场上销售的医疗器械始终维持其安全性和有效性。那么,FDA究竟如何进行市场监督?是否会进行抽查?答案是肯定的,而且其监督方式远不止于“抽查”那么简单。以下是FDA主要的市
在当今**化的商业环境中,企业社会责任和供应链透明度日益成为企业发展的关键要素。Sedex验厂作为一种广泛认可的供应链审核方式,帮助众多企业提升管理水平、增强市场竞争力。许多企业在考虑进行Sedex验厂时,首先关心的往往是费用问题。实际上,Sedex验厂的成本并非固定不变,它受到多种因素的影响,包括企业规模、行业特点、现有管理基础以及审核的具体要求等。影响Sedex验厂成本的主要因素企业规模是决定
在当今建设项目日益增多的环境下,选择一家合适的造价咨询公司显得尤为重要。对于需要专业服务的客户而言,如何从众多选择中找到*匹配的合作伙伴,是一个值得深入探讨的话题。专业能力是首要考量因素一家优秀的造价咨询公司应当具备全面的专业服务能力。从项目建议书及可行性研究,到经济评价报告的编审,再到项目概预算编审和设计方案比选,这些基础服务能力都是衡量公司专业水平的重要标准。优秀的造价咨询公司还能提供优化设计
在当今快速变化的商业环境中,企业持续追求卓越运营已成为保持竞争力的关键要素。随着数字化转型浪潮席卷各行各业,如何将传统管理方法与新兴技术**结合,成为众多组织关注的焦点。精益数字化培训正是在这样的背景下应运而生,为企业提供了一条通向*运营的清晰路径。精益数字化培训融合了经典管理理念与先进数字技术,旨在帮助企业构建更加高效、透明的运营体系。这种培训方式不仅保留了传统管理方法的精髓,更通过数字化工具
在现代企业管理中,质量管控已成为企业运营的核心环节。随着市场竞争的日益激烈,企业如何通过高效的流程优化和人才培养来提升整体运营水平,成为众多管理者关注的焦点。六西格玛作为一种先进的管理方法,不仅强调流程的精益化,更注重通过系统化的培训和咨询,帮助企业培养高素质的质量管控人才,从而实现可持续的发展。六西格玛管理方法源于对流程稳定性和质量一致性的追求,其核心理念是通过数据驱动的分析和改进,减少流程中的
在当今竞争激烈的市场环境中,企业如何通过高效的管理方法实现持续改进和永续经营,已成为一个关键议题。合规性精益生产作为一种结合精益理念与行业标准的管理模式,正逐渐成为众多企业提升运营效率、优化资源配置的重要工具。它不仅强调消除浪费、提高生产力,还注重与行业规范相融合,确保企业在合规的前提下实现可持续发展。精益生产的核心理念与合规性要求精益生产源于对生产流程的深度优化,其核心在于通过系统化的方法识别并










